
Theoretical and Computational Chemistry
Accurate simulations of molecules and materials have the potential to greatly acellerate the development of new technologies (better solar cells, batteries, drugs, etc.). On a theoretical level, we know how to do these simulations exactly. The problem? Running these simulations for any interesting molecule or material would take longer than the heat death of the universe. The main issue is accurately modeling the electrons, which has come to be known as the electronic structure problem. The entire electornic structure field is focused on developing fast, accurate approximations to the true quantum mechanical simulations. I believe the most promising solution to this problem lies in the development of both physics-informed machine learning models and fast (linear-scaling) algorithms to apply these models to large systems.
Featured Work
Linear Scaling Hybrid Density Functional Theory
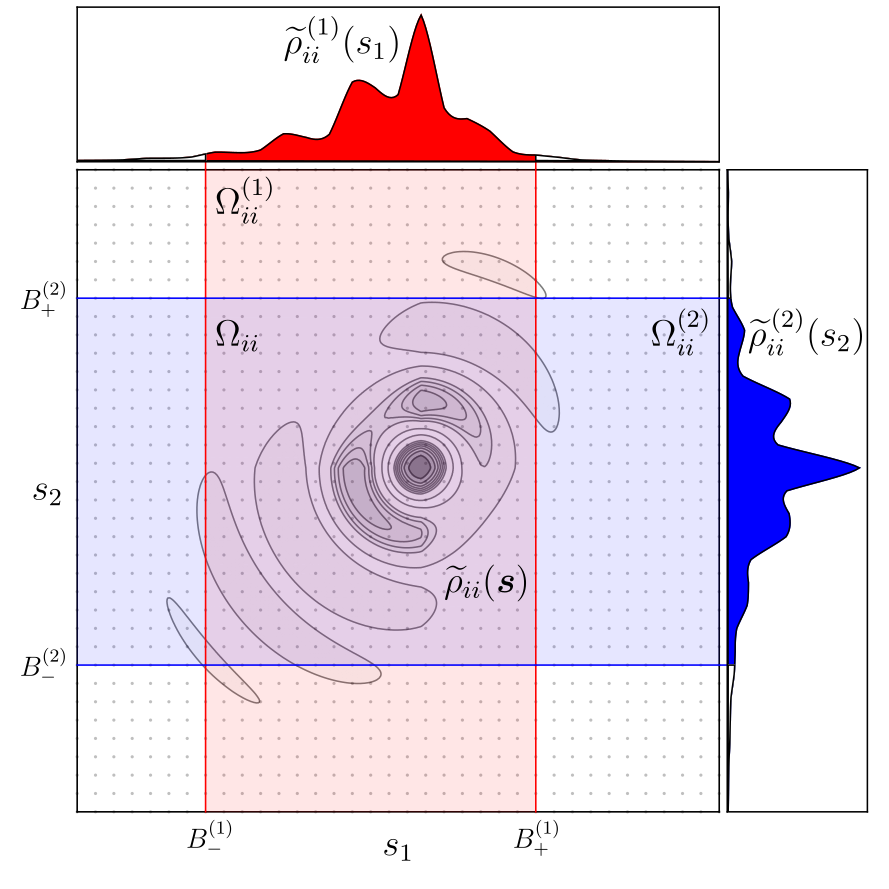
Density Functional Theory (DFT) is the standard method for studying chemical and physical systems computationally. Approximations within DFT are generally accurate enough to get useful results, while still maintaining a tolerable computational cost for many systems. But, sometimes the chemistry/physics is complex, and you need so-called hybrid DFT. The key component to hybrid DFT is known as exact exchange, which is typically quite costly to compute when compared to simpler approximations. As a result, studying large systems with hybrid DFT was historically just a pipe-dream. With this goal, we’ve developed an exact exchange algorithm that scales linearly with system size, ultimately reducing the cost of hybrid DFT to be comparable to standard DFT. My main contribution is the development of an algorithm that is straightforwardly applicable to a wide variety of heterogeneous systems, and with a predictable and controllable level of accuracy. Paper and code release coming soon!
AI Polymer Design

One of my favorite recent projects was developing an ML model dubbed the PolyEthylene Property Predictor (PEPPr) to predict the properties of HDPE given the molecular weight distribution (the distribution of molecule sizes) of the sample. Polymers are difficult to understand from first principles because of their stochastic nature, making ML an attractive tool for this problem. Leveraging synthetic data augmentation, we were able to train a model that is stable enough to guide the design of polymers with a desired set of properties, resulting in the experimental synthesis of stronger, easier to process materials. Additionally, we demonstrated how to use PEPPr to overcome some of the problems associated with material degredation during the recycling process.
Uniting Physical and Empirical Models
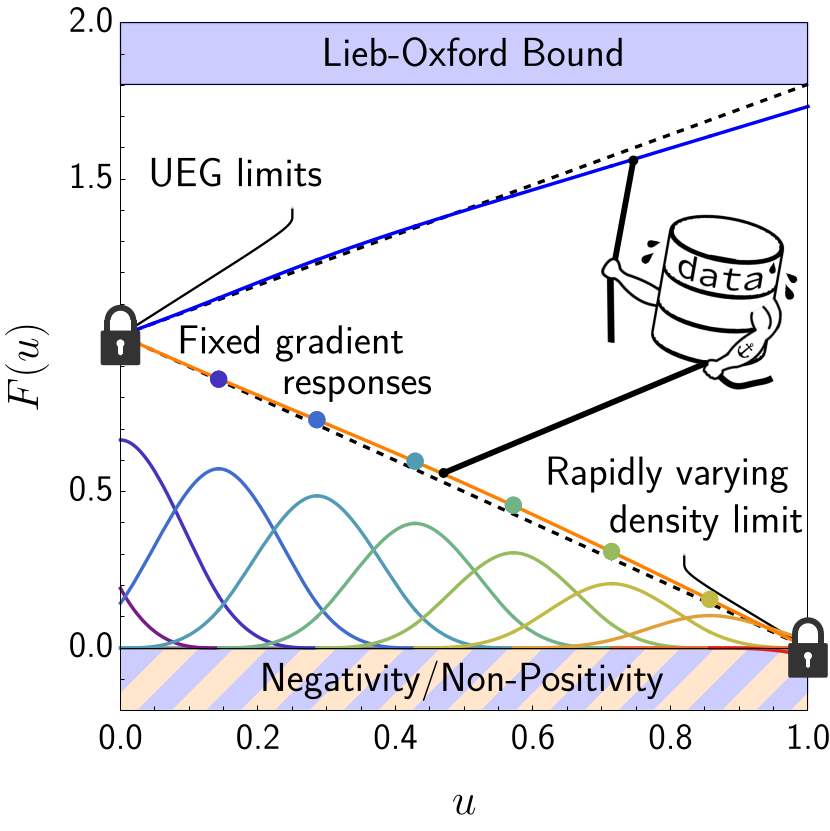
My first foray into training my own empirical quantum chemical approximation followed the realization that most other empirical methods ignored many of the underlying fundamental physics, hoping to learn such information directly from the data. I realized that the vast majority of this physics can be written in terms of constraints and regularization penalties, effectively filling in the gap between the empirical and non-empirical approximations. Using this technique, we showed that an approximation that uses both data and physical constraints outperforms some of the most common approximations still used today.
Chemical Database Construction

My first Ph.D. project was to construct and analyze a database of close-range molecule-molecule interactions. At the time of publishing, this was the largest benchmark-quality database, containing about 8,000 unique interactions. My analysis had a number of key insights into some of the most commonly used approximations, most notably how the error made in a simulation scales with respect to system size in a manner that resembles a bias-variance breakdown, even for non-empirical approximations. The resulting publication was selected as the issue cover article and won Editor’s choice for 2021 in the Journal of Chemical Physics.
Publications
- Z. M. Sparrow, H.-Y. Ko, J. Zhang, and R. A. DiStasio Jr., Robust Linear-Scaling Hybrid Density Functional Theory with Controllable Accuracy for Finite-Gap Condensed-Phase Systems, in preparation.
- Z. M. Sparrow, B. G. Ernst, R. A. DiStasio Jr., Benchmarking Wavefunction Theory and Density Functional Approximations for Short-Range Non-Covalent Interactions, in preparation.
- Z. M. Sparrow, R. Kang, B. G. Ernst, R. A. DiStasio Jr., Decomposing Conformational Energy Errors into Geometric and Potential Energy Curvature Components: Application to NECE-2026, in preparation.
- R. Kang, Z. M. Sparrow, B. G. Ernst, R. A. DiStasio Jr., NECE-2026. A Large Benchmark Database of Non-Equilibrium Conformational Energies, in preparation.
- H.-Y. Ko, J. Zhang, M. F. C. Andrade, Z. M. Sparrow, S. Ghosh, P. T. Lin, R. A. DiStasio Jr., Reactive Transport of Water Ions: Presolvated Proton Donors behind the Scenes, in review.
- Z. Shen, Y. Yang, Z. M. Sparrow, B. G. Ernst, T. K. Quady, R. Kang, J. Lee, Y. Yang, L. Tu and R. A. DiStasio Jr., Learning Molecular Conformational Energies Using Semi-Local Density Fingerprints, J. Phys. Chem. Lett., 51 16 (2025). Link
- J. Hu, Z. M. Sparrow, B. G. Ernst, S. M. Mattes, G. W. Coates, R. A. DiStasio Jr., B. P. Fors, Designing Polymers with Molecular Weight Distribution Based Machine Learning, J. Am. Chem. Soc., 147 12 (2025). Link
- H.-Y. Ko, M. F. C. Andrade, Z. M. Sparrow, and R. A. DiStasio Jr., High-Throughput Condensed-Phase Hybrid Density Functional Theory for Large-Scale Finite-Gap Systems: The SeA Approach, J. Chem. Theory Comput., 19 13 (2023). Link
- Z. M. Sparrow, B. G. Ernst, T. K. Quady, and R. A. DiStasio Jr., Uniting Non-Empirical and Empirical Density Functional Approximation Strategies Using Constraint-Based Regularization, J. Phys. Chem. Lett., 13 30 (2022). Link
- V. Prabhakaran, J. Romo, A. Bhattarai, K. George, Z. M. Norberg1, D. Kalb, E. Aprà, E., P. A. Kottke, A. G. Fedorov, P. Z. El-Khoury, G. E. Johnson, and J. Laskin, Integrated photoelectrochemical energy storage cells prepared by benchtop ion soft landing, Chem. Commun., 58 9060 (2022). Link
- Z. M. Sparrow, B. G. Ernst, P. T. Joo, K. U. Lao, and R. A. DiStasio Jr., NENCI-2021 Part I: A Large Benchmark Database of Non-Equilibrium Non-Covalent Interactions Emphasizing Close Intermolecular Contacts, J. Chem. Phys., 155 184303 (2021). Cover Article & Editor’s Choice 2021 Link
- M. Pearce, Z. M. Sparrow, T. R. Mabote, and R. Sanchez-Gonzalez; stoBEST: An Efficient Algorithm for Increased Spatial Resolution in Two-Component Molecular Tagging Velocimetry, Meas. Sci. Technol. 32 035302 (2021). Link
- Y. Vidavsky, M. R. Buche, Z. M. Sparrow, X. Zhang, S. J. Yang, R. A. DiStasio Jr., and M. N. Silberstein, Tuning the Mechanical Properties of Metallopolymers via Ligand Interactions: A Combined Experimental and Theoretical Study, Macromolecules 53 2021 (2020). Link
Talks & Presentations
- Z. M. Sparrow, R. Kang, B. G. Ernst, R. M. Hendley, R. A. DiStasio Jr., Construction and Analysis of the NECE-2024 Benchmark Non-Equilibrium Conformational Energy Database, Institute for Pure and Applied Mathematics Advancing Quantum Mechanics with Mathematics and Statistics Reunion Conference 2, Lake Arrowhead, CA. (2024).
- Z. M. Sparrow, H.-Y. Ko, Ju-an Zhang, and R. A. DiStasio Jr., Robust Linear-Scaling Hybrid Density Functional Theory with Controllable Accuracy for Finite-Gap Condensed-Phase Systems, Institute for Pure and Applied Mathematics Advancing Quantum Mechanics with Mathematics and Statistics Reunion Conference 1, Lake Arrowhead, CA. (2023).
- Z. M. Sparrow, H.-Y. Ko, and R. A. DiStasio Jr., Towards Routine Use of Hybrid DFT for Large-Scale Finite-Gap Condensed-Phase Systems, Institute for Pure and Applied Mathematics Advancing Quantum Mechanics with Mathematics and Statistics Seminar Series, Los Angeles, CA. (2022).
- Z. M. Sparrow, Predicting Chemical Properties on Your Computer Using Data and Physics, Chemistry Department Seminar, St. Olaf College, Northfield, MN. (2022).
- Z. M. Sparrow, B. G. Ernst, T. K. Quady, and R. A. DiStasio Jr., Uniting non-empirical and semi-empirical density functional approximation strategies using constraint-based regularization, American Chemical Society Spring Meeting, San Diego, CA. (2022).
- Z. M. Sparrow, B. G. Ernst, P. T. Joo, K. U. Lao, R. A. DiStasio Jr., NENCI-2021: A Large Benchmark Database of Non-Equilibrium Non-Covalent Interactions Emphasizing Close Intermolecular Contacts, American Physical Society March Meeting, Online. (2021).
- Z. M. Sparrow, B. G. Ernst, P. T. Joo, K. U. Lao, R. A. DiStasio Jr., NENCI-2020: A Large Benchmark Database of Non-Equilibrium Non-Covalent Interactions with an Emphasis on Close Contacts, American Physical Society March Meeting, Denver, CO. (2020) (cancelled)
- Z. M. Norberg1, M. Pearce, S. Hoops, O. Dmytrenko, M. Richey, and R. Sanchez-Gonzalez, An Efficient Algorithm for Increased Spatial Resolution in Two-Component Molecular Tagging Velocimetry, National Conference on Undergraduate Research, Memphis, TN. (2017)
- Z. M. Norberg1, V. Prabhakaran, J. Laskin, Fabrication and Optimization of an Integrated Photoelectrochemical Device Using Ion Soft-Landing, Pacific Northwest National Laboratory Summer Research Symposium, Richland, WA. (2016)
Academic Profiles
Google Scholar: Zachary M. Sparrow
ORCiD: 0000-0001-6163-2843